INPUT
- Reference sequence :
- - In order to run the BLAST with query and references, you need to put reference sequences. If you have the user-defined amino acid or nucleotide sequences, you can upload it on the system. Otherwise, you can put the accesion number such as NC_000000.For more information, please refer NCBI site
- Input File Format :
- - Input file is an aassembled contig. The format should be "FASTA". The AGORA allows only one assembled query contig. For more information about FASTA, please refer to NCBI site
- Type :
- - Select Choloroplast or Mitochondrion for your organellar genome
- Genetic Code
- - This code is used for running the tBLASTn
- Standard
- Vertebrate Mitochondrial
- Yeast Mitochondrial
- Mold Mitochondriali
- Invertebrate Mitochondrial
- Ciliate Nuclear
- Echinoderm Mitochondrial
- Euplotid Nuclear
- Bacteria and Archaea
- Alternative Yeast Nuclear
- Ascidian Mitochondrial
- Flatworm Mitochondrial
- Blepharisma Macronuclear
- For more information about genetic code please see NCBI
- Maximum number of matched sub gene's count per each contig :
OUTPUT
- Sample output is here
- Output :
- - As you see below examples, output file is the BLAST result that includes amino acid and nucloetide. The Query is set to the refereces and Data base is set to query. The number of matched position is decided upon the "Maximum matched sub gene's count"
-
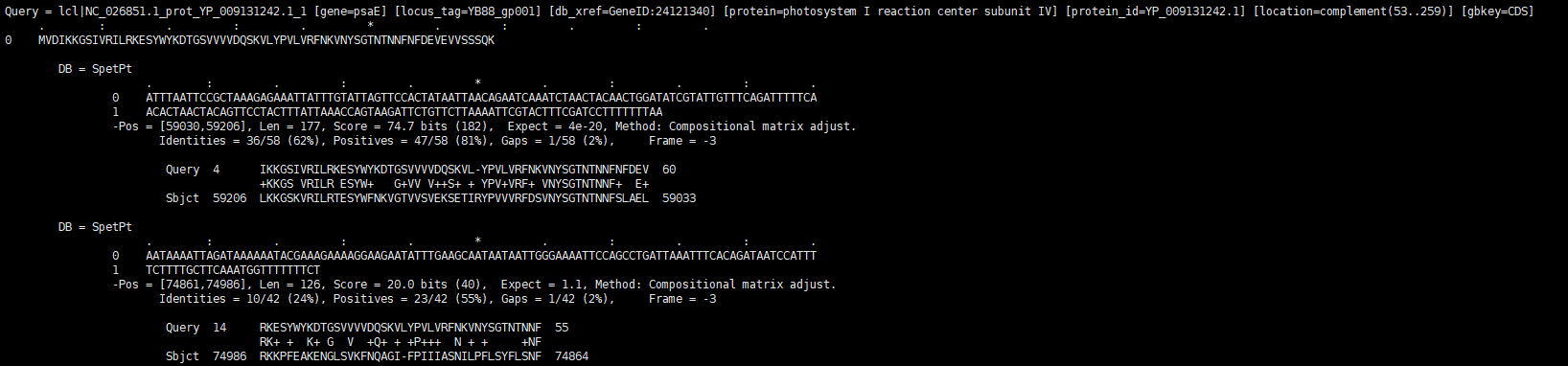 The blast result of amino acid
The blast result of amino acid -
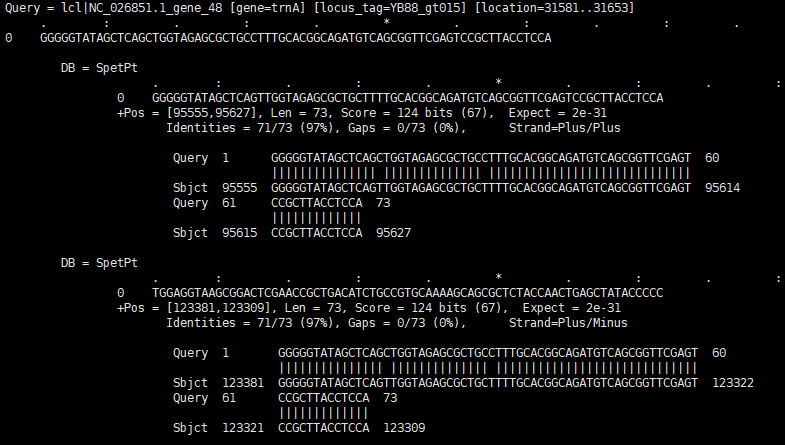 The blast result of nucleotide
The blast result of nucleotide - Amino acid db sequences :
- - This file includes the amino acid data base sequences. If the user uploaded the user-defined sequence, this file is same to that uploaded file. Otherwise, system is automatically generated from the NCBI.
- Amino acid sequences :
- - This is CDS translation files that is matched from the BLAST
- output CSV file :
- - This file provides the start and end position, direction and gene product for each gene.
- Nucleotide db sequences :
- - This file is nucleotide data base sequences.
- Nucleotide sequences :
- -The FASTA formatted seuqneces file is includes the BLAST mached sequences.
- GenBank File format :
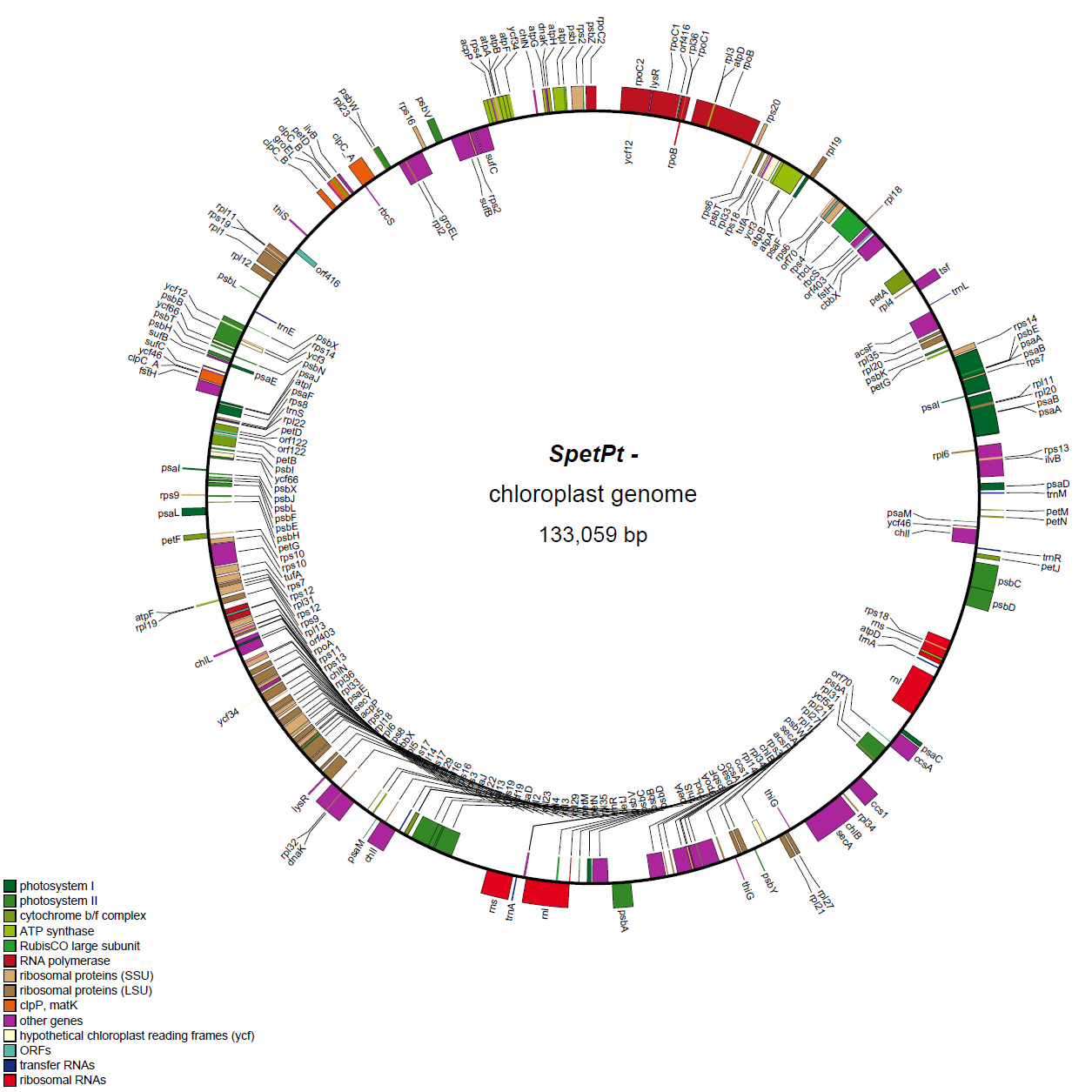
- - This file is GenBank formatted file. With this file we draw the circular gene map by running OGDRAW
- OGDRAW :
- - If all genes are matched correctly, you can see the figure. Here is example